The actin cytoskeleton appears to play two essential roles in the activation of the T-mobile. One of those functions is the progression and movement of the shape of the T-mobile, adding to the formation of the immune synapse. The other is the formation of a scaffold for signaling. Components. This review focuses on the recent convergence of mobile biology and immunology studies with the role of the actin cytoskeleton in creating the molecular basis for immune synapse formation and T-motile signaling.
More than 20 years ago, studies on ligand-induced movement of immunoglobulins on the surface of B cells drew attention to a specific dating between the actin cytoskeleton and antigen receptors1,2,3. Recent studies on T cells have refocused our attention on this specific dating. Actin filaments play at least two roles in antigen recognition. The first considers microscale molecular movements on the surface of T4,5 cells. This large-scale molecular rearrangement has effects on the formation of an immunological synapse arranged in a distinct supramolecular activation. The current role of actin filaments appears to involve signaling complexes that depend on a scaffold of actin filaments7,8,9,10,11,12. The actin scaffold can also recruit or stabilize specialized membrane domain names enriched with glycolipids and signaling molecules. involved in T13,14,15 activation.
The formation of the immunological synapse is a multistep process that begins with adhesion between the T cell and antigen presenting cell (APC). Theoretically, it is possible that the T cell antigen receptor (TCR) could initiate this process by interacting with major histocompatibility complex molecule (MHC)-peptide complexes, but in practice this would require too many MHC-peptide complexes and too much time16,17. Instead, the initial adhesion is mediated by integrins such as LFA-1 or by non-integrin molecules such as CD2-CD58 or DCSIGN–ICAM-318,19,20,21. The common goal of these adhesion mechanisms is to overcome the barrier to close cell-cell contact posed by the negatively charged glycocalyx of the T cell and APC22,23. Both the T cell and APC have large glycocalyx components such as the mucin CD43, which come into conflict with each other at a ∼50–100 nm separation of cell membranes24. This distance cannot be spanned by the TCR and MHC-peptide complex, which interact at ∼15 nm25,26. One solution to this problem is to use abundant adhesion molecules—such as CD2 and CD58—to bring cells to within 15 nm of each other, by formation of thousands of transient, low affinity interactions23,27,28,29,30. T cells are sensitive to small numbers of MHC-peptide complexes on the APC31. Precise alignment of apposing membranes at ∼15 nm is probably essential for this high sensitivity. However, small adhesion molecules such as CD2 and CD58 are also prevented by the glycocalyx from interacting and require active processes to initiate adhesion32. The integrin LFA-1 and its major immunoglobulin superfamily ligand ICAM-1 can interact at ∼40 nm and can thereby initiate adhesive interactions when appropriately activated33,34. These large adhesion molecules initiate active mechanisms to promote the interaction of smaller adhesion molecules, bringing the actin cytoskeleton to center stage.
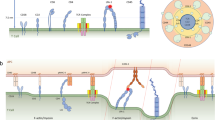
The actin cytoskeleton plays a dual role in regulation of LFA-1. The resting leukocyte has two surface domains: low flat surfaces and microvilli. Integrins such as LFA-1 are located on the low flat surface along with other large glycoproteins like CD4335 (Fig. 1). In contrast, L-selectin is located at the ends of the microvilli where they are well positioned to initiate interactions with ligands on endothelial cells36. The flat surface is supported by a cortical actin cytoskeleton that is interlaced with microtubules and intermediate filaments37. LFA-1 on resting leukocytes is maintained in a low activity state by an inhibitory interaction with the cortical actin cytoskeleton38,39,40. This inhibitory interaction may be mediated in part by talin, a large actin and integrin binding protein41. The inhibitory interaction prevents lateral movement of the integrins in the membrane that may be important for encountering ligands and clustering of integrins. In this state the lymphocyte is not well prepared for antigen recognition and requires non-antigen dependent signals to rearrange its surface topology for high sensitivity to MHC-peptide complexes.
Conversion of a spherical resting T cell to a polarized migratory T cell. Actin is in green, microtubules in blue and myosin II in red. Right arrows indicate movements of cell surface structures on the lymphocytes that have just been exposed to a chemokine gradient (Δ). Chemokine receptor on naïve T cells include CCR7, which binds SLC and ELC; and CXCR4, which binds SDF-1. The microvilli are swept to the back of the cell along with CD43. Arrows on left hand diagram indicate direct of force exerted by actin polymerization on the leading edge.
Activation of lymphocytes releases the inhibitory cytoskeletal interaction, so that LFA-1 can diffuse in the plane of the membrane38. At this stage it is likely that activating signals are non-antigen-specific signals from chemokines or other environmental cues that link the preparation for antigen recognition to specific environments (for example, lymph nodes) or processes (for example, inflammation)42,43. Chemokine signaling activates a heterotrimeric G-protein–initiated signaling cascade that activates phospholipase C, phosphatidylinositol-3-kinase, protein kinase C and Rho family G-proteins leading to activation of myosin light chain kinase and local actin polymerization44. Activation of myosin II, the conventional form of myosin that forms bipolar “thick filaments”, initially increases the cortical stiffness of the leukocyte by contracting the cortical actin shell45. Further contraction by myosin II results in the collapse of the original cortical actin cytoskeleton to the side of the cell occupied by the microtubule organizing center (Fig. 1). This retraction of the resting cell cortical actin cytoskeleton creates an opening for new actin polymerization to form membrane protrusions at the leading edge of the newly polarized and motile lymphocyte. The myosin II and actin aggregate in the new trailing edge along with the microvilli and a number of proteins, including the large glycocalyx component CD43, through its interaction with the adapter protein ezrin46. The movement of CD43 to the rear edge of the cell may promote detachment of adhesion at the rear of the cells, while promoting interactions at the leading edge by relieving steric inhibition. The activation-induced polarization of the lymphocyte is functionally important. Lymphocytes are more sensitive to MHC-peptide complexes or anti-TCR antibody-induced activation at the leading edge than at the trailing edge47. Although initial polarization is stimulated by chemokines, long-term exposure to cytokines such as interleukin 2 maintains the polarized phenotype, even in the absence of external chemotactic gradients48.
Several state-of-the-art houses contribute to effective receptor-ligand interactions. New actin polymerization is concentrated in the main protrusions and the receptors in these regions are primarily cellular49. The actin structures in these regions also respond to receptor engagement. For example, the interaction of a cellular integrin with a ligand causes a new adhesion-stabilizing interaction with the actin cytoskeleton50. It has been noted that this positive interaction would possibly be mediated by α-actinin, an integrin and an actin-binding protein other than talin41. Talin may also be involved at this level by stabilizing LFA-1 clusters and the higher affinity form of LFA-151,52. Cytoskeletal interactions with engaged integrins cause expansion of close contact zones53. Integrin clusters are also transported in a directed manner, resulting in the movement of T cells over the substrate or APC54,55. The areas adjacent to the active integrin clusters are forced to come into close contact with the adjacent membrane5. These spaces of closed and forced membrane apposition are most likely the nucleation sites for the interaction between smaller adhesion mechanisms and antigen receptors, and can also be characterized simply as touch sensory domains.
The generation of sensory contact domains probably involves formation of actin-based protrusions, such as filipodia (thin spikes) or lamellipodia (flat sheets) (Fig. 1). These protrusions generate force based on actin polymerization56. The protrusive force exerted by these structures is counter-balanced by the anchoring force of the integrin interactions at the center of the contact area5. The formation of actin protrusions is controlled by small G-proteins of the Rho family including Cdc42 and RhoA57. RhoA is inactivated by the C3 ribosyltransferase. The C3 ribosyltransferase blocks LFA-1 activation and also inhibits production of interleukin 2 and sustained Ca2+ elevation in response to TCR engagement58,59,60. An efficient scheme for moving antigen receptors to within ligand binding range of the apposing membrane would be to nucleate actin assembly from the antigen receptors themselves. A link between the TCR and actin-based structures may be formed by VASP, an actin filament–coupling protein. One mechanism for linking VASP to the TCR is through the adapter protein Fyb that interacts with Fyn and VASP61,62. There are probably other mechanisms for forming this linkage as T cells from Fyn-deficient mice have normal sensitivity to antigen63. VASP is associated with protrusive structures employed in initial interactions of epithelial cells and thus may provide an appropriate linkage to actin to initiate TCR engagement with ligands64. Regardless of this, the first critical role for the actin cytoskeleton appears to be collaboration with chemokine receptor–mediated signals and adhesion molecules to initiate physiological TCR engagement.
When MHC-peptide agonist complexes are placed on an antigen-delivering surface with integrin ligands, the coordination of adhesion, the actin cytoskeleton, and antigen receptors takes on another character. The T cell stops its migration and generates a central zone of integrin engagement surrounded by a region of close contact that includes maximally engaged TCR5 (Fig. 2a). Over a few minutes, the engaged TCRs are transported to the center of the contact zone and the engaged integrins are forced into a surrounding ring (Fig. 2b) 5,6. This trend can be robust for several hours and sustained signaling over this time scale is required for full T cell activation (Fig. 2c)67. TCR monitoring on the surface of T cells shows an initial bipolar distribution. Small clusters form at the nascent synapse in reaction to the interaction of the MHC-peptide complex, with a giant cluster at the opposite pole of the cell. This paradoxical clustering at the cell pole far from the APC is likely greatest due to myosin II activation and actin filament contraction at the original trailing edge (see web videos 1-4; http://dx. doi . org/10. 1038/20000000). Over a period of several minutes, small groups of TCRs reorganize to form a large central organization within the immunological synapse and additional TCRs move from the distal pole to the central organization of the synapse. Transport of receptors to the immunological synapse was discovered by Wülfing and Davis and requires myosin II4. Therefore, in the T cell, myosin II participates in the generation of actin at the back of the cell, which is reversed upon synapse formation. This polarity reversal of actin movement requires interaction of the TCR with LFA-1 or CD284. The configuration of the organization of immunological synapses depends on the density and quality of the MHC-peptide complexes5,6. Therefore, the immunological synapse formation procedure reaches the entire surface of T cells and the actin and myosin cytoskeletal systems to translate the short-lived interaction of TCR and MHC-peptide complexes into a robust supramolecular structure. to the proportions of the length of a cell phone.
(a–b) Schematic style for the configuration of immunological synapses connected to the main points of actin dynamics. MHC-peptide complexes are green and ICAM-1 is red. Insets refer to inserts containing a detailed description of the molecules that bind the TCR and actin at other stages of synapse formation. (a) Finishritic nucleation style of actin polymerization triggered by activation of the Arp2/3 complex in the presence of actin (light parallelogram, bound to ATP; purple parallelogram, bound to ADP), capping protein (red square), and cofilin (blue square). triangles). Initially, ATP-actin is added to the barbed end of the filament, but is then hydrolyzed to ADP + inorganic phosphate. Once this happens, the Arp2/3 complex dissociates. Cofilin then promotes inorganic phosphate dissociation leading to filament fragmentation. Phosphorylation of cofilin via LIM kinase inhibits actin binding and increases filament half-life. (b) The role of myosin II bipolar filaments (red) in contracting the actin network to shape the core cluster of the engaged TCR. Formation of long-capped actin filaments will likely require cofilin inhibition. One prospective mechanism is the ability of the Rac spin form of p21 spin kinase to activate LIM kinase, which phosphorylates cofilin and inhibits its function. These more robust filaments can then be connected to TCR-associated actin via actin filament cross-linking proteins such as α-actinin or L-plastin. Actin gel contraction through the effects of myosin II on TCR core cluster formation. (c) The contracted dfinishritic actin network serves as a scaffold for the kinesis protein C-θ and other molecules involved in the sustained signaling process. It is assumed that express adapters or scaffolding proteins are required for this process.
The TCR is a multisubunit transmembrane glycoprotein composed of the antigen-binding α and β subunits, as well as the invariant CD3 complex and ζ chains. The cytoplasmic domain names of CD3 and ζ polypeptides involve immune receptor tyrosine activation motifs (ITAMs) that are phosphorylated by src family circle tyrosine kinases, such as Lck, upon TCR engagement. Each ζ subunit has 3 ITAMs and each ITAM has two tyrosines. ITAMs are required for TCR68-induced actin polymerization. The ITAM phosphorylation patterns on the ζ polypeptide are ordered such that phosphorylation of any of the tyrosines on an ITAM is only observed with MHC-peptide agonist complexes69. ITAMs with either phosphorylated tyrosine are required to recruit the non-receptor tyrosine kinase ZAP-70 via their paired SH2 domain names70. ZAP-70 activation can be inhibited by the phosphatases SHP-1 and CD45 and by the degradation sites c-Cbl and Cbl-b71,72,73,74,75,76. The giant transmembrane tyrosine phosphatase CD45 also plays a positive role in Lck activation. These positive and negative regulators help establish thresholds for T-mobile activation. ZAP-70 phosphorylates sites leading to the recruitment of the adapter proteins SLP-76 and LAT77,78,79,80. Regarding the actin cytoskeleton, SLP-76 is unusual in that it recruits Vav and Nck11. Nck recruits WASp (the deficiency of which causes Wiscott-Aldrich syndrome). Vav activates the Rho circle of the G proteins Cdc42 and Rac. In its inactive state, WASp assumes a closed conformation that cannot interact with the key actin regulator Arp2/3, a complex involving actin-related proteins 2 and 3, as well as five other polypeptides81. Activated Cdc42 interacts with WASp to induce an open conformation that recruits and activates the Arp2/3 complex. Inositol phospholipids, generated in part through the action of phosphatidylinositol 3-kinase linked to the TCR-recruited adapter molecule LAT, also appear to contribute to WASp activation through interaction with the pleckstrin homology domain of WASp80. . 82. WASp also affiliates with a proline-rich adapter protein, WIP, which has actin and profilin binding sites and increases actin polymerization83. The activated configuration of Rho family circle G proteins orchestrates the formation of complex actin-based structures through the activation of multiple effector molecules57. Activated Cdc42 generates filipodia, thin projections involving actin, and Rac generates lamellipodia, flat spaces of active actin polymerization in membranes that can appear as “ruffles” at the outer edge of the immunological synapse (see Web Movies 5-7; http:/ /dx. doi. org/10. 1038/20000000). In T mobiles, these signaling processes, assessed by tyrosine phosphorylation and Ca2+ mobilization, are maximal a few minutes after the initial interaction with the APC, when the immunological synapse forms.
Key molecular parts recruited to the activated TCR are sufficient to induce dramatic actin polymerization. Recruitment of Nck and N-WASp to vaccinia virus debris in the cytoplasm of inflamed cells stimulates the formation of actin “rockets” that propel the debris through the cytoplasm in a procedure that depends solely on actin polymerization84. . The movement of Vaccinia debris into the cytoplasm is similar to that of Listeria monocytogenes, in which the bacterial surface protein ActA directly recruits Arp2/3 to induce actin spindle formation85,86. Listeria-based motility has now been reconstituted into a formula with 4 purified proteins: actin, Arp2/3, capping protein and cofilin87. This study was an important milestone because it provided formal evidence that actin polymerization generates a mobile force. The mechanism of actin polymerization is thought to involve the ability of activated Arp2/3 complexes to bridge the sides of existing actin filaments and nucleate new ends of barbed filaments (the rapidly growing end) while capping the pointed end88. . This “dendritic nucleation” procedure produces an expanding network of polymerized actin that exerts a force on the particle89. The ability of WASp to stimulate this procedure has been directly observed in vitro82.
Vaccinia utilizes a tyrosine phosphorylated transmembrane protein to recruit Nck and N-WASp and is thus more analogous than Listeria to the antigen receptor systems. In T cells, mutations in SLP-76 or Nck result in impaired TCR-stimulated actin polymerization11. Mice deficient in Vav1 or WASp show specific defects in T cell activation and actin polymerization in response to antibody cross-linking9,10,90. Human patients deficient in WASp exhibit defects in T cell activation and actin-based structures91. These defects were observed despite the presence of two other Vav isoforms and at least one additional WASp isoform. The initial TCR clusters that form at the periphery of the nascent immunological synapse are likely to be sites of extensive dendritic nucleation growth of actin networks (Fig. 2a, inset). The immunological synapse model predicts that synapse formation will be at least partially defective in Vav- and WASp-deficient mice.
The close sensory contacts adjacent to the integrin participation sites are likely generated through polymerization of membrane-nucleated actin via the Arp2/3 complex. The surface regions of active protrusion-rich T cells are more sensitive to MHC-peptide complexes in APCs. or anti-CD3 bead cells47,65. Therefore, the bumps should be TCR-rich. TCR has low lateral mobility on the surface of T cells, and only 20% of proteins show loose diffusion. Therefore, it is very likely that the TCR has a basal arrangement with the cytoskeleton that may be mediated simply by proteins such as Fyn, Fyb, and VASP, as described above. The TCR would then be forced onto the surface of the APC to allow effective interaction with the MCH-peptide surface complexes of the APC.
Following antigen receptor triggering, it is clear that the TCR recruits Nck, WASp and Vav in a manner that is likely to activate the Arp2/3 complex and trigger rapid actin polymerization (Fig. 2a, inset). This polymerization may drive the MHC–peptide-engaged TCR clusters further against the APC surface to expand the area of close contact.
The mechanism by which the MHC-peptide–engaged TCR are then transported to the center of the contact to form the central TCR cluster is not known. A likely mechanism would involve the activation of myosin II. Myosin II can be activated by the interaction of Ca2+-calmodulin with myosin light chain kinase, which phosphorylates the regulatory light chain of myosin92. The bipolar myosin filaments then contract the actin filaments into a dense gel bringing along associated surface proteins (Fig. 2b, inset). This mechanism is thought to account for the flow of actin towards the rear end of cells undergoing amoeboid motion93,94,95. Stabilization of the actin filaments to provide a substrate for the myosin II based motility may be promoted by activation of the p21 activated kinase (PAK) at the TCR through the action of Vav and SLP-7611. Activated PAK in turn activates LIM kinase, which phosphorylates an inhibitory residue on cofilin96. Cofilin participates in the disassembly of actin filaments. Inhibition of cofilin should result in a longer half-life of actin filaments that could then be contracted by activated myosin II. An alternative mechanism for engaged TCR movement to the center of the synapse would be that actin polymerization itself would drive the complexes inward. Either of these hypotheses can account for the observation that the number of MHC-peptide complexes in the central compartment of the immunological synapse is related to the degree of complete ITAM phosphorylation and ZAP-70 recruitment. This is because both models are sensitive to the rate of actin filament formation stimulated at the TCR in response to ZAP-70 recruitment5,69.
A desirable facet of the immunological synapse is the interdependence and coordination of adhesion and the formation of signaling complexes. The engaged TCR and related actin filaments move toward the middle of the synapse, but LFA-1, which is also functionally related to actin, remains in a discrete peripheral adhesion ring. This happens even when the LFA-1 ligand, ICAM-1, moves freely. Therefore, substrate anchoring through ICAM-1 cannot explain the fact that LFA-1 does not also cluster in the medium. One conceivable mechanism is the arrangement of LFA-1 with talin (a giant cytoskeletal protein with multiple integrin binding sites), the cytoskeletal protein vinculin, and actin filaments. Talin is highly accumulated in spaces of LFA-1 participation within the immunological synapse6. Talin has been implicated in the lateral immobilization of integrin complexes41 and would possibly play a vital role in integrin-mediated protective contacts against scanning toward the middle of the synapse. The adhesion molecule CD2 also appears to identify and maintain a position outside the center of the synapse, but largely within the integrin ring5. This localization of CD2 can be stabilized through CD2AP, an adapter protein involved in the movement of CD2 and the formation of strong cell-cell junctions in the kidney97,98.
The formation of a solid synapse in the inductive phase of an immune reaction can serve several functions. For example, a migratory T-mobile will have to avoid moving when it encounters an antigen. The synapse would allow the T-motile to concentrate cytokine secretion on a PCA or mobile target99. The synapse provides a structural “hardware” solution for integrating signs from the environment in the form of supramolecular compartments. It appears that those compartments solidify for several hours and that a threshold number of MHC-peptide complexes (around 50) will have to work in parallel to generate the structure. Therefore, this massively parallel activation procedure is well suited for producing a high-fidelity deterministic sign based on the number and quality of the MHC peptide as well as the adhesion molecules in the APC (Fig. 3(a). The configuration of these supramolecular devices can, as it should, measure the amount of MHC peptides and the interaction kinetics with the TCR and thus delineate a rigorous threshold for complete T-mobile activation.
( a ) The parallel mode of triggering the effects of the TCR from the incorporation of the activated TCR in the core group. The star symbolizes signage. Data on the downregulation of TCRs recommend that TCRs are occupied at other times through each MHC-peptide complex. However, many TCRs are triggered in parallel by incorporating into a higher-order structure. The entire group of TCRs is degraded en bloc when the T cells and APCs are separated. 119(b) The serial mode of triggering the effects of the TCR from the immediate dispersal of the triggered TCR without incorporation into a higher-order structure. MHC-peptide complexes can be used to interact with many serial TCRs.
In the past, a different mode of T-cell signaling based on serial compromise has been proposed. 100In this procedure, TCRs are activated for signaling and then removed at the single receptor (Fig. 3 (b). Molecular segregation at tactile velocities provides a co-receptor-independent mechanism for signaling initiated through single TCR engagement events 101,102,103. However, serial activation has not been directly observed in cell contacts and is studied indirectly through the downregulation of TCR, a procedure that is also consistent with serial activation or solution of an immunological synapse. Serial activation is also supported by studies in which T cells move during signaling, including from APC to APC7,104. Although in those examples significant movement precludes the formation of a robust immune synapse, the molecular organization of those cellular synapses has not been studied. Direct serial activation would require following the history of single TCR molecules, which is likely to be readable soon given the current speed of advances in imaging technology105,106.
Serial engagement is thought to require actin polymerization to identify contact zones and appropriate signaling scaffolds (but this is not mandatory for large-scale receptor movement)7,107. This signaling mode can achieve maximum sensitivity because each MHC-peptide complex can cause many TCR108. Such a formula would likely get a significant number of incorrect mobile activation occasions due to the challenge of separating basic spontaneous signals from actual interaction occasions. In the first activation of mature T cells, the misactivation load may also be very high (e. g. autoimmunity), so the increased constancy through synapse formation may also simply be an evolutionary reaction to this. carry. On the contrary, in T-mobile development, the accusation of incorrect activation in positive and negative format might be less problematic. Incorrect activation in positive variety has effects on the production of a mature T mobile that poorly recognizes self-MHC and is therefore not vulnerable to peripheral activation through a self-MHC-peptide complex. Most likely, these mobile phones die due to lack of survival signals in the periphery109. Incorrect activation has several negative effects on the loss of a mobile that would have been a useful mature T mobile. A small number of these errors may not be serious because they do not generate destructive motives.
Therefore, some signals for T-mobile progression and, in all likelihood, survival of mature T-mobiles would possibly be gained in a serial rather than parallel manner. Serial interaction predicts that the signaling procedure in immature T-mobiles is tuned to distinguish between the noise of spontaneous receptor activation and a very small number of remote interaction events. This is a “soft integration” process that relies on determining the rate and duration of individual TCR interaction events, as opposed to the “hard integration” process of the immunological synapse, where a robust design is formed to contain Parallel TCR signaling events. . It has been proposed that CD4 or CD8 lineage decisions could be controlled through the duration of signaling in immature T mobiles110, and that the role of CD4 in promoting synapse formation5 could provide long-term signaling (approximately 10 h) necessary for an immature lineage. T Mobile. expressing CD4 and CD8 to expand into mature CD4+ T cells. Therefore, it will be very vital to what extent other types and progressive stages of T mobiles depend on those chosen signaling mechanisms, in parallel or in series, to respond to MHC-peptide complexes in vivo.
The molecular mechanisms linking the activation of mature T-mobiles to synapse formation are unclear. Most likely, there are differences in signaling pathways between immature and mature T-mobiles. Specific synapse-dependent signaling molecules appear in key facets of mature T-mobiles. Mobile activation. For example, protein kinase C-θ (PKC-θ) is required for NF-κB activation in mature T-mobiles, but not in immature T-mobiles111. While it is conceivable that this developmental difference in PKC-θ needs is due to some redundancy in immature T-mobiles, it is also conceivable that this difference reflects a genuine difference in signaling mechanisms. PKC-θ is unique among the PKC bureaucracy in its localization in the central supramolecular activation group within the immunological synapse112.
Disruption of actin filaments blocks T cell activation immediately7,8,113. One hypothesis for this effect is that actin filaments establish a scaffold for assembly of signaling complexes. Adapter proteins would be involved in linking specific signaling pathways to actin as has been demonstrated in other systems114,115. One candidate for recruitment through the actin scaffold is PKC-θ, the kinase recruited to the center of the mature synapse (Fig. 2c, inset). PKC-θ activity is dependent on Vav, but its recruitment to the TCR cluster appears to be indirect and is dependent on the ability of Vav to induce actin polymerization12. The actin scaffold may also recruit or stabilize glycolipid-rich rafts that concentrated in the vicinity of the engaged TCR14. The impact of the actin scaffold may be to sustain signaling complexes by protecting components from degraadation. For example, CD28 co-engagement with the TCR is required for effective raft recruitment in primary stimulation of mature T cells. Cbl-b is a ring finger protein that functions in suppression of signaling by acting as a ubiquitin ligase to target neighboring proteins for degradation by the proteosome73,74. The Cbl-b−/− mouse has a hyper-responsive T cell phenotype that can be alleviated by crossing to the CD28−/− mouse. One could speculate that the role of CD28 in organizing the cytoskeleton in the immunological synapse may be, in part, to protect certain signaling molecules from proteolysis by linking them to the actin scaffold, and thus prolonging signals from engaged TCR. The contribution of the actin scaffold to sustained signaling is probably important for full T cell activation which can require up to 20 h of sustained signaling67. The kinetics of APC clusters with T cells in vivo are also consistent with an important role of sustained signaling in vivo116,117.
Since Mechnikoff’s earlier experiments on amiobocyte attack on foreign bodies,118 immunologists have appreciated the importance of motility and cytoarchitecture in the immune response. Our current knowledge of the actin cytoskeleton is based on data accumulated from a large number of style formulas as varied as the biochemistry of soil amoebas, the genetics of yeasts, and the microscopy of fish keratocytes. Advances in protein chemistry, mammalian genetics, and imaging technologies have made it possible to apply lessons learned from these gendered formulas to express cytoskeletal proteins in T lymphocytes associated with antigen-presenting formulas in vitro or in the whole-organism context. . The ability to formulate and test hypotheses based on actin mechanisms shared by various organisms should lead to further identification and discovery of specific aspects of the lymphocyte cytoskeleton signaling formula that could provide immunological treatments for the future.
We would like to thank D. Schafer, K. Blumer and A. I would like to thank Mr. Shaw for his advice and comments. MLD and J. A. C. they are supported by grants from the National Institutes of Health.
Department of Pathology and Immunology, Washington University School of Medicine, 660 S. Euclid Ave. , St. Louis, 63110, MO, United States
Michael L. Dustin
Department of Cell Biology and Physiology, Washington University School of Medicine, 660 S. Euclid Ave., St. Louis, 63110, MO, USA
John A. Cooper
Initial distribution of the T cell receptor. T cell receptors tagged with Alexa-488 H57 Fab were imaged by confocal microscope (30 x time lapse). Note the non homogeneous distribution of the TCR on the surface and the evidence of membrane dynamics. Images captured with Shannon Bromely, Kenneth Johnson and John Heuser.
Formation of an immunological synapse. Mobile T receivers were classified with Alexa 488-H57 Fab and mobile receivers were symbolized under a confocal microscope (30x time-lapse). This TCR 3A9 transgenic T-mobile has just come into contact with an egg-pulsed B-mobile C3F6 (BPA). White lysozyme. The first symbol is a glowing symbol showing mobile T at the bottom right and mobile B at the top left. The film spans a generation of 8 minutes in 20 seconds of video. At the end of the movie, the TCR moved to the center of the touch area. Some relief in general fluorescence is due to photobleaching. Images captured with Shannon Bromely, Kenneth Johnson and John Heuser.
Three dimensional rendering of early time point from movie 2.
Three-dimensional representation of the vanquished moment from the film 2.
Integrin mediated sensory contact. Interference reflection image of a T cell contact with a serum coated supported planar bilayer. Image is shown in real time.
Contact area dynamics in immunological synapse. T cell reacting with planar bialyer containing MHC-peptide complex and serum proteins (integrin ligands). Note membrane ruffling at the periphery of the immunological synapse. Image is shown in real time. Images captured with Cenk Sumen.
Contact domain dynamics triggered only through TCR participation. The mobile T reacts with a planar bilayer containing only the MHC-peptide complex. Note deep membrane dynamics, which would possibly reflect stimulation of polymerization through action through the transiently engaged TCR. Dynamic and irregular appearance. The symbol is displayed in real time. Images captured with Chistophe Wülfing.
Reprints and permissions
Date of publication: July 2000
DOI: https://doi.org/10.1038/76877
Anyone you share the following link with will be able to read this content:
Sorry, there are no shareable links for this article lately.